Un modelo matemático identifica genes resistentes a fármacos contra el cáncer
Un equipo científico de la Universidad de Málaga ha diseñado un algoritmo que indica qué células dentro de un mismo tumor presentan una mayor sensibilidad a ciertos fármacos frente a otras que oponen más resistencia.
Los resultados del trabajo ayudarán a investigar enfoques terapéuticos más precisos.
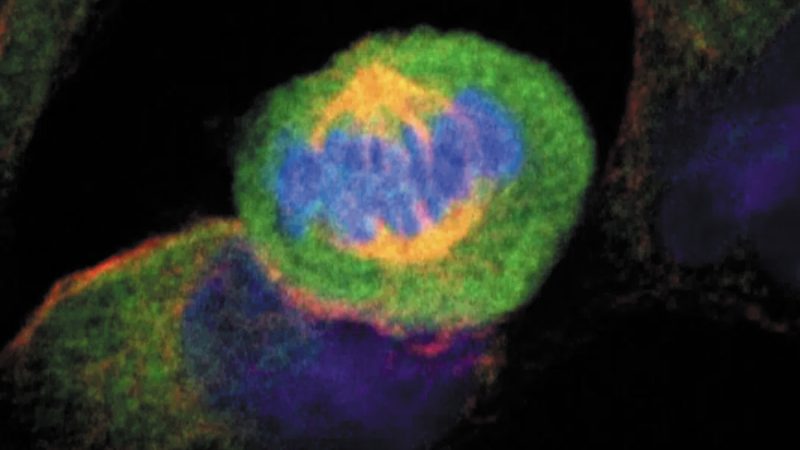
Científicos del Departamento de Biología Molecular y Bioquímica de la Universidad de Málaga han desarrollado un modelo matemático que identifica genes resistentes a fármacos contra el cáncer.
El estudio, publicado en la revista Molecular Genetics and Genomics, abre nuevas vías de abordaje con estrategias que detectan los genes de resistencia a medicamentos específicos en cada uno de los subtipos celulares que componen el tumor.
Los investigadores han observado dentro de una misma población de líneas celulares cómo algunas células cancerígenas rechazan la acción de la quimioterapia y otras presentan mayor sensibilidad a ella. Los resultados del trabajo ayudarán a investigar nuevos enfoques terapéuticos más precisos, dirigidos a los subtipos celulares con mejor disposición a cada uno de los distintos tratamientos.
Hasta el momento, los investigadores observaban las diferencias en la expresión génica del conjunto de células que conforman la muestra poblacional total del tumor. Pero el nuevo sistema permite la combinación de los datos de la composición en tipos celulares y de los valores de expresión de distintas líneas cancerígenas con el fin de encontrar los genes responsables de la resistencia a la medicación, expresados de forma específica en las distintas subpoblaciones celulares del tumor.
En esta línea, los expertos exponen el nuevo modelo de cálculo que ha desarrollado las observaciones sobre 16 líneas tumorales, ocho resistentes y ocho sensibles al tratamiento con uno de los medicamentos más usados para quimioterapia en los cánceres más frecuentes.
A través del modelo que proponen los expertos, usado frecuentemente en el estudio de terremotos o en óptica, se mejora la identificación de biomarcadores que indican la resistencia a la quimioterapia teniendo en cuenta que dentro de cada población celular existe una heterogeneidad en la manera en la que expresan su respuesta.
Por tanto, se trata de reconocer cuántas subpoblaciones celulares se encuentran dentro de esas líneas y caracterizarlas en función de su contribución a la expresión de los genes que causan la respuesta ante los medicamentos.
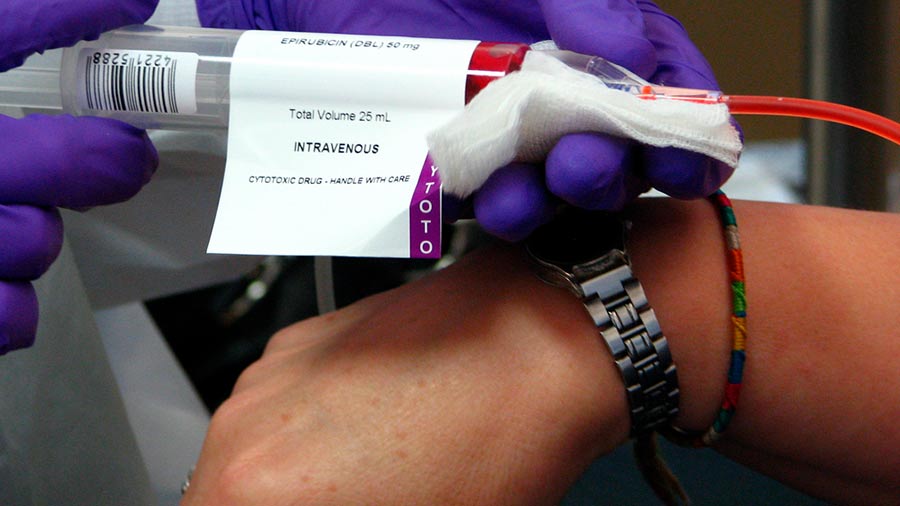
“Uno de los mayores problemas en la lucha contra tumores es la resistencia a fármacos, bien presente de una manera innata en el tumor o adquirida tras la exposición a la quimioterapia. La preocupación por desarrollar estrategias de tratamiento eficaces nos ha llevado a elaborar una fórmula que explique las causas genéticas de esta resistencia debidas a la composición en distintos tipos celulares de la población tumoral”, indica Juan Antonio García Ranea, autor del artículo.
“Expresión génica de subpoblaciones celulares”
Tradicionalmente, los algoritmos utilizados para calcular las dosis necesarias en el tratamiento contra el cáncer se fundamentaban en los datos teóricos. Sin embargo, las situaciones clínicas concretas son totalmente diferentes a las condiciones genéricas. Por otro lado, los algoritmos basados en modelos matemáticos necesitan menos medidas experimentales para ajustar las cantidades requeridas.
Para solventar estos obstáculos y ajustarse más a la realidad de los casos, el sistema que proponen los autores se apoya en el análisis y observación de la expresión génica de subpoblaciones celulares mediante deconvolución. Este es un modelo matemático basado en la regresión lineal múltiple, que consiste en la restauración de datos que han sido degradados. Es decir, permite combinar distinta información con el fin de obtener los datos iniciales y plantear las conclusiones sobre los orígenes o causas en unos y otros casos.
Los expertos parten de una población celular en la que se incluyen subpoblaciones resistentes a los tratamientos y otras que no lo son. La deconvolución permite el análisis del desarrollo evolutivo de la expresión de genes en estas subpoblaciones de manera diferenciada, determinando por qué y en qué medida unas desarrollan resistencia a un medicamento que se usa normalmente en quimioterapia y otras no lo hacen.
Hasta el momento, los valores obtenidos de un tumor eran valores medios de la expresión de los distintos tipos celulares que lo componen, aunque éstos no son reflejo exacto de la realidad heterogénea del tumor, ya que las técnicas actuales no permiten observar la expresión génica en cada tipo celular. Sin embargo, la deconvolución posibilita la descomposición de las señales medias para obtener valores que sean realmente representativos de los subtipos celulares y relacionarlos con el desarrollo de la resistencia.
En el estudio se llegaron a identificar casi 90 genes específicos de los distintos subtipos celulares, cuya expresión diferencial permanecía oculta dentro de los valores de expresión medios del tumor. Los autores demostraron en el trabajo que dichos genes se comportan como biomarcadores capaces de distinguir entre líneas resistentes y sensibles a la medicación en una muestra dispar de tipos de cáncer.
Por tanto, una línea futura en la que los investigadores plantean continuar sus estudios orientados hacia la identificación de los posibles mecanismos y sistemas moleculares implicados en la resistencia, asociados a los nuevos genes identificados.
El equipo de trabajo forma parte de la red europea de excelencia denominada ‘Systems Microscopy NoE’ y el proyecto ha sido financiado por la comisión europea de investigación dentro del VII Programa Marco.
Fuente: SINC