¿El coronavirus está mutando?
Liliana Rojo
Doctorado en Ciencias en el Uso, Manejo y Preservación de los Recursos Naturales por el CIBNOR
La respuesta corta es sí, y el rastreo de las mutaciones permite inferir información que puede ser clave para el manejo adecuado de la pandemia.
Vamos poniendo en contexto, los virus son partículas de ARN o ADN (información genética) envuelto en una capa de proteínas, en el caso del SARS-CoV-2, es ARN. Los bloques se constituyen el ARN, se llaman nucleótidos y están dispuestos en tripletes llamados codones que proporcionan el código para construir aminoácidos, los bloques que forman las proteínas del virus. Una mutación es un cambio en la información genética, es decir, en uno de estos nucleótidos del RNA del virus. SARS-CoV-2 tiene ~29,811 nucleótidos y como todos los virus, sufre de manera natural y espontanea mutaciones, aunque la mayoría de estas mutaciones no causan efectos en la biología del virus, no alteran los síntomas que producen, ni su capacidad infecciosa.
Es posible rastrear las mutaciones de SARS-CoV-2 gracias al enorme esfuerzo de científicos de casi todo el mundo que han depositado secuencias genéticas en bases de datos como GISAID, una iniciativa que promueve el acceso global a una enorme cantidad de información genética, datos clínicos y epidemiológicos de virus de influenza y otros relacionados como SARS-CoV-2.
GISAID alberga mas de 30,000 secuencias de genomas de SARS-Cov-2 (mayo-26). A partir de esta información, plataformas como Nextstrain y Cov-GLUE han desarrollado herramientas que permiten analizar la diversidad nucleotídica, es decir, los sitos que son diferentes entre las secuencias reportadas, estas diferencias son causada por mutaciones y su análisis permite visualizar la estructura geográfica y patrones de propagación de SARS-CoV-2. Se observa que algunas variantes tienen una clara distribución geográfica (ver Figura 1, construida en https://nextstrain.org/ncov/global).
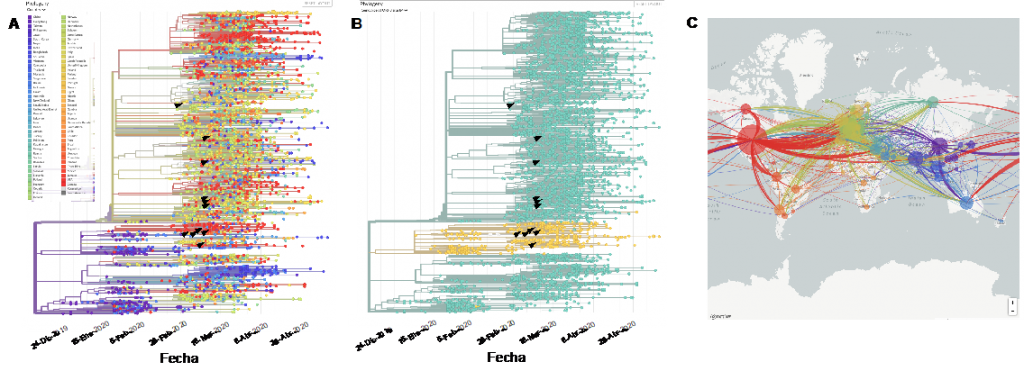
En la plataforma Nexstrain también es posible visualizar los índices de variabilidad dentro de los genomas reportados. Es notable que ciertos sitios son mas propensos a presentar cambios, detectados principalmente en regiones que contienen el código para las proteínas de la envoltura viral, como la proteína S (spike) y la N (nucleocapside). Estas diferencias en un solo nucleótido (SNPs) tienen el potencial de funcionar como marcadores para la definición de subtipos de SARS-CoV-2. Caracterizar y cuantificar la presencia de tipos o subtipos virales en poblaciones específicas permiten monitorear la prevalencia de variantes, sus patrones de transmisión e incluso virulencia y pueden ser clave para generar una respuesta rápida y manejo adecuado de epidemias como la del coronavirus.
Hasta ahora no se ha confirmado la presencia de variantes genéticas de SARS-Cov-2 con mutaciones que les confieran mayor virulencia, aunque algunos estudios han sugerido que sí, con pocas evidencias experimentales, por ejemplo, el 3 de marzo se publicó un articulo liderado por el Dr. Jian Lu, de la Peking University en Beijing, en donde se identifican dos subtipos principales de SARS-CoV-2 con base en dos cambios puntuales encontrados en 103 secuencias analizadas, los autores los denominaron como tipos S y L, reportando una mayor prevalencia del subtipo L en la provincia de Hubei, China, los autores sugieren que el subtipo L es mas agresivo y se dispersa más rápidamente. Aunque esta aseveración ha generado controversia, pues otros científicos aseguran que el trabajo de Lu presenta limitaciones metodológicas y sobre-interpretación. Para llegar a la conclusión que presenta Lu es necesario mostrar mucha más evidencia experimental, además la prevalencia de dichas variantes es comúnmente generada por azar debido a un fenómeno conocido como efecto fundador y no por presión selectiva, como es sugerido por Lu y colaboradores.
Lo que sí podemos saber, de acuerdo a los datos depositados en GISAID es que la variante L está presente en aproximadamente el 89% de los genomas reportados, ambas variantes se han detectado en México, país sub-representado en esta base de datos con 27 genomas reportados, Brasil ha depositado 95 genomas y Chile 139 (junio-2). En ese contexto, al no tener suficiente información sobre las variantes genéticas que se distribuyen en nuestro país se prescinde de datos valiosos para la evaluación del estatus y control de la epidemia.
Fuente: CIBNOR