Desentrañan el misterio del Síndrome de Andersen Tawil tipo 1
Unos científicos han logrado desvelar la causa de las arritmias y la muerte súbita en la enfermedad llamada Síndrome de Andersen-Tawil tipo 1. Esta es una dolencia rara de herencia autosómica dominante caracterizada por frecuentes arritmias ventriculares, asociadas a alteraciones del desarrollo de grado muy variable.
Los científicos que han hecho el hallazgo son de dos equipos del Centro Nacional de Investigaciones Cardiovasculares (CNIC) en España.
Dirigidos por los doctores José Jalife y Juan Antonio Bernal, los investigadores han descubierto una función fundamental previamente desconocida de los canales Kir2.1, que controlan las propiedades eléctricas esenciales de células excitables como las del músculo cardíaco, las del músculo esquelético y las neuronas.
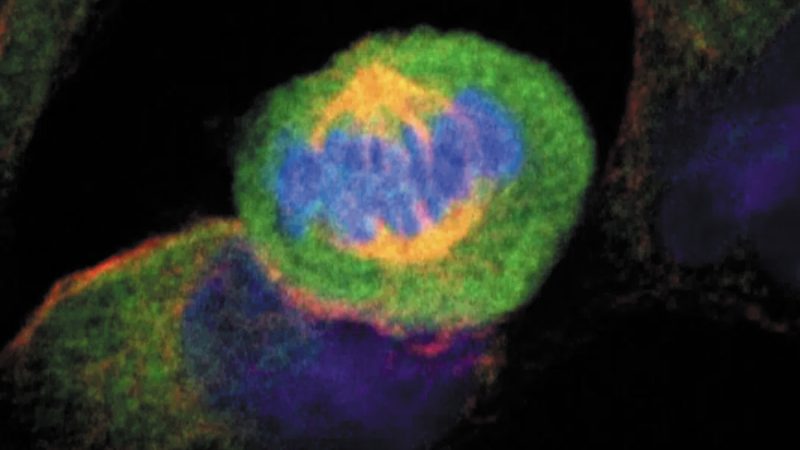
Gracias a una combinación de imágenes de microscopía confocal, y de ensayos bioquímicos y electrofisiológicos, estos equipos han desvelado una doble localización y función de los canales Kir2.1 controlando los flujos de potasio a través de las membranas celular y endoplásmica, estando involucrados en el control de la actividad eléctrica y el acoplamiento de excitación-contracción, respectivamente.
En el estudio, los equipos del CNIC han demostrado que estos dos distintos microdominios de proteína Kir2.1 están presentes en diferentes especies y en diferentes células musculares, lo que indica que sustentan funciones relevantes conservadas y generalizadas en las células.
Además, tal como señala el Dr. Bernal, en el estudio también se ha hecho una caracterización detallada de un nuevo modelo de ratón de la enfermedad.
La inyección intravenosa en el animal vivo de virus adenoasociados (AAV) que contenían un gen mutante KCNJ2, codificador de una proteína Kir2.1 deficiente en tráfico a su localización fisiológica en las células del corazón, ayudó a los autores del estudio encontrar una nueva explicación a nivel molecular de las arritmias potencialmente mortales que ocurren en pacientes con ATS1.
“Dichas arritmias se solapan con las arritmias de otra enfermedad hereditaria igualmente maligna llamada taquicardia ventricular polimórfica catecolaminérgica (CPVT)”, explica el Dr. Jalife.
En un estudio de revisión de resultados de otras investigaciones, realizado recientemente por parte del mismo equipo y publicado en la revista académica Cardiovascular Research, el ATS1 se manifiesta como una tríada de síntomas incluyendo arritmias ventriculares, parálisis periódica y rasgos dismórficos producidos por mutaciones de pérdida de función en KCNJ2, el gen que codifica la proteína Kir2.1, pero los mecanismos de las arritmias han sido desconocidos hasta ahora.
“El modelo de ratón -explica el Dr. Álvaro Macías, primer autor del nuevo estudio- recapitula las anomalías eléctricas cardíacas encontradas en pacientes ATS1, incluida la alta carga de arritmia y la susceptibilidad a la taquicardia/fibrilación ventricular (TV/FV) responsable de la muerte súbita”.
Los datos presentados ahora proponen un nuevo mecanismo molecular potencial para explicar las arritmias, la debilidad del músculo esquelético y la parálisis periódica reportadas en pacientes con ATS1.
“Es importante destacar que también demuestran que el tratamiento con flecainida, un fármaco comúnmente utilizado en el ámbito clínico y que en ocasiones falla en controlar las arritmias en pacientes con ATS1, exacerba sustancialmente el fenotipo ATS1 y aumenta la posibilidad de arritmias en el modelo de ratón”, añade el Dr. Jalife.
Estos últimos datos, continúa Jalife, “advierten a los clínicos sobre el uso de este fármaco que, dependiendo del caso específico, podría ser perjudicial para los pacientes con esta enfermedad”.
En resumen, la función dual de Kir2.1 debería conducir a terapias novedosas y más eficaces para ATS1 y otros trastornos superpuestos relacionados con alteraciones de la dinámica del calcio, ya sean hereditarios como la CPVT, pero posiblemente también adquiridos como la insuficiencia cardiaca que afecta a millones de personas en todo el mundo.
El nuevo estudio se titula “Kir2.1 dysfunction at the sarcolemma and the sarcoplasmic reticulum causes arrhythmias in a mouse model of Andersen–Tawil syndrome type 1”. Y se ha publicado en la revista a académica Nature Cardiovascular Research.
Fuente: noticiasdelaciencia.com