
Jessica Nayelli Sánchez Carranza
La Dra. Jessica Nayelli Sánchez Carranza estudió la carrera de Químico Industrial en la Facultad de Ciencias Químicas e Ingeniería de la UAEM y la Maestría y Doctorado en la Facultad de Farmacia. Actualmente es Profesora Investigadora de Tiempo Completo de la Facultad de Farmacia UAEM y miembro de la Academia de Ciencias de Morelos, A.C.
Esta publicación fue revisada por el comité editorial de la Academia de Ciencias de Morelos.
¿Alguna vez te has preguntado por qué un medicamento puede ser altamente efectivo para una persona, pero no tener ningún efecto en otra, o incluso causar efectos secundarios graves? La respuesta a este enigma puede encontrarse en las variaciones en nuestros genes.
¿Qué es la Farmacogenética?
La farmacogenética estudia cómo las variaciones genéticas (polimorfismos) afectan la respuesta a los fármacos. Un polimorfismo de nucleótido único (SNP) es la variación de una secuencia de ADN que ocurre cuando se altera un solo nucleótido (adenina, timina, citosina o guanina). Los SNP suelen presentarse en al menos el 1% de la población, y pueden influir en cómo se absorben, distribuyen, metabolizan y eliminan los fármacos en el cuerpo.
La importancia de los Genes en la Respuesta a los Fármacos
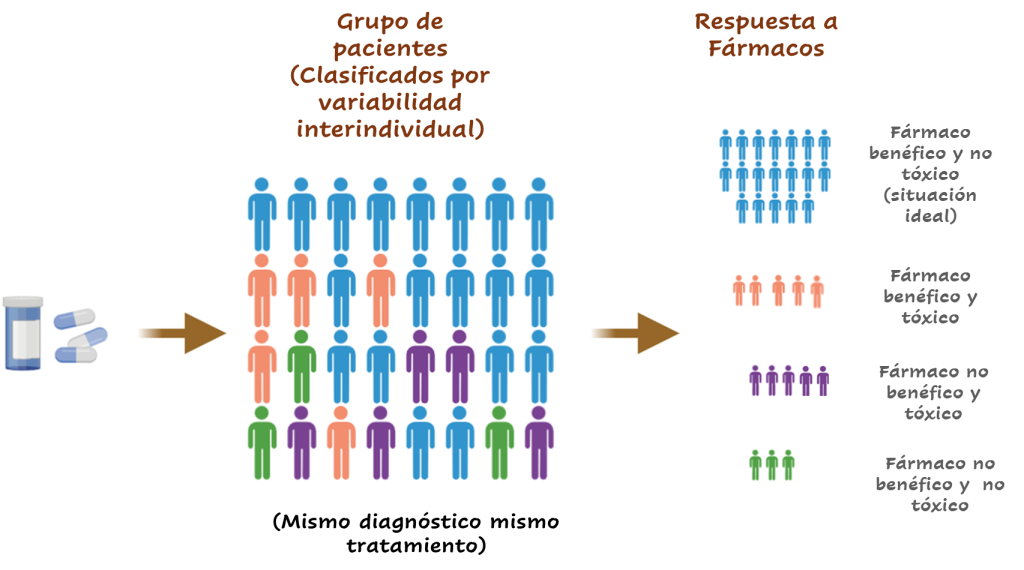
Muchos genes codifican proteínas conocidas como enzimas. Estas tienen innumerables funciones, incluida la descomposición (metabolismo) de los fármacos. Aunque puede sonar extraño que hayan enzimas que modifiquen nuestros medicamentos, en realidad no es tan raro. Evolutivamente, nuestro organismo ha tenido que defenderse de diferentes agentes tóxicos en el ambiente y los medicamentos son identificados como un agente tóxico. Las personas que no responden a los fármacos como se espera pueden tener variantes genéticas que alteran la función y/ó expresión de una enzima que actúa sobre el fármaco, provocando que el metabolismo de un fármaco sea demasiado rápido, demasiado lento, o que no se metabolice, por lo que una dosis prescrita puede tener poco o ningún efecto en el tratamiento del paciente y/ó provocar efectos secundarios importantes. La figura 1 es una representación de un estudio farmacogenético, donde un grupo de pacientes con el mismo diagnóstico pueden presentar diferentes respuestas a un mismo tratamiento: fármaco benéfico y no tóxico (situación ideal), fármaco benéfico y tóxico, fármaco no benéfico y tóxico, fármaco no benéfico y no tóxico.
Enzimas involucradas en el metabolismo de fármacos
Los genes de la familia del citocromo P450 (CYP450) son cruciales para el metabolismo oxidativo de los xenobióticos, que son compuestos químicos no producidos por el organismo, como fármacos y toxinas. Esta familia de genes codifica enzimas responsables del metabolismo de alrededor del 80% de los fármacos disponibles. Este sistema enzimático no está compuesto por una única enzima, sino por una amplia familia de hemoproteínas. Hasta la fecha, se han identificado más de 2000 isoformas diferentes del citocromo P-450.
A menudo, los pacientes presentan determinadas variaciones en estos genes, que afectan la velocidad con la que se metabolizan los fármacos, lo que influye en su eficacia y seguridad. En la figura 2 se muestra la clasificación de los cuatro posibles tipos de metabolizadores de fármacos, con base en el número y tipo de variantes funcionales presentes en los genes involucrados en el metabolismo.
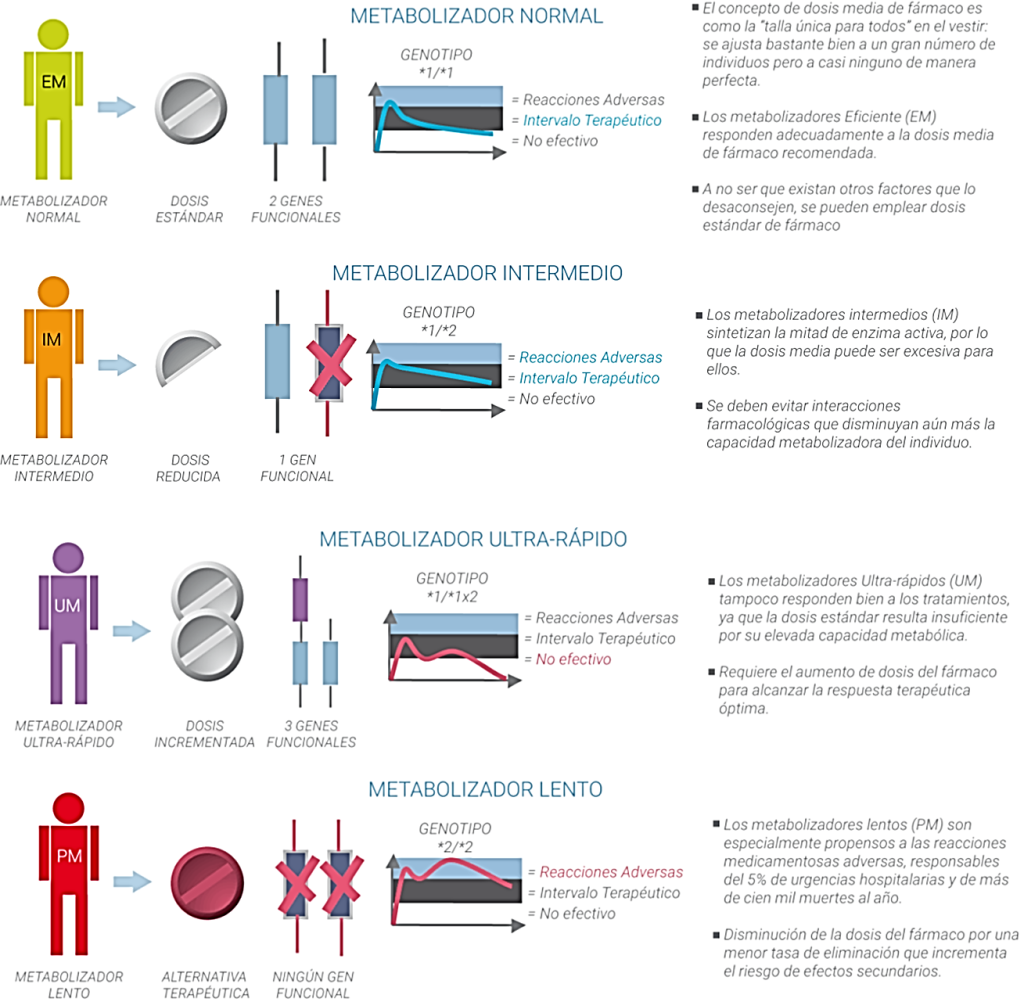
Como puede verse en la Fig. 2, solo los metabolizadores normales tendrán una buena respuesta al medicamento en las dosis comúnmente empleadas. Los metabolizadores ultrarápidos, al poder modificar mejor el medicamento, requerirán dosis mayores de él para tener una respuesta adecuada. Por el contrario, los metabolizadores intermedios o los lentos, corren el riesgo de una reacción adversa, requiriendo una menor dosis del fármaco. Desde luego, conocer de antemano a qué grupo pertenece un paciente permitirá reducir riesgos al administrar un medicamento.
Un ejemplo de estas situaciones podemos encontrarlo con las variantes de CYP2D6, responsable del metabolismo de muchos fármacos comúnmente prescritos, incluidos antipsicóticos, analgésicos, betabloqueantes y antidepresivos tricíclicos como la amitriptilina.
Los individuos cuyas variantes en CYP2D6 los hacen metabolizadores lentos de amitriptilina, con una dosis estándar tendrán niveles plasmáticos más altos de este antidepresivo, en comparación con los metabolizadores normales. Debido a que la dosis estándar de amitriptilina puede provocar un mayor riesgo de eventos adversos en estas personas, se recomienda evitar el uso de amitriptilina u otros antidepresivos tricíclicos y considerar el uso de un fármaco alternativo. Alternativamente, se considerará una reducción del 50% de la dosis inicial recomendada, con un seguimiento estrecho de su efecto (Figura 2).
Aquellas personas con más de dos copias funcionales de CYP2D6 son metabolizadores ultrarrápidos (Figura 2). En estas personas, el aumento en el metabolismo de la amitriptilina conduce a una menor disponibilidad del fármaco activo y a una respuesta terapéutica deficiente. Por lo que, debido a la posible falta de eficacia, se recomienda considerar un fármaco alternativo a la amitriptilina que no sea metabolizado por CYP2D6. Si se justifica su uso, se recomienda aumentar la dosis inicial y monitorización terapéutica del fármaco para guiar los ajustes de dosis.
Variantes farmacogenéticas implicadas en la diferente respuesta a Fármacos
- CYP2D6 y Codeína
CYP2D6 es un gen particularmente complejo, debido a la gran cantidad de variantes que presenta, incluyendo pérdidas y ganancias de copias. Como se mencionó anteriormente, CYP2D6 desempeña un papel crucial en el metabolismo de aproximadamente el 20% de los fármacos que se recetan comúnmente. Algunos de estos fármacos incluyen antidepresivos, antipsicóticos, analgésicos y fármacos anticancerígenos. Uno de los opiáceos principales que se metabolizan a través de esta enzima y que cuentan con mayor evidencia es la codeína, un profármaco que es modificado por nuestro organismo para producir el metabolito activo, la morfina. Sin embargo, la actividad de la enzima puede variar según el genotipo del individuo, lo que se traduce en diferentes tipos de metabolizadores:
Metabolizadores normales: Tienen dos copias funcionales del gen CYP2D6. Metabolizan la codeína de manera eficiente. Los pacientes experimentan un efecto analgésico adecuado a dosis estándar de codeína.
Metabolizadores ultrarrápidos: Tienen múltiples copias funcionales del gen. Metabolizan rápidamente la codeína a morfina. De tal manera que hay un aumento de la concentración de morfina, lo cual puede llevar a estos pacientes a alcanzar concentraciones plasmáticas del metabolito tan elevadas que pongan en riesgo su vida.
Metabolizadores lentos: Tienen dos copias no funcionales del gen CYP2D6. Su capacidad para convertir la codeína en morfina es limitada. Los pacientes como resultado presentan concentraciones plasmáticas de morfina mucho menores. Esto puede llevar a un alivio insuficiente o incluso nulo del dolor, además de una acumulación del fármaco en el organismo y un mayor riesgo de toxicidad.
Por su parte el tramadol es un analgésico que se utiliza para tratar el dolor moderado a intenso en adultos y en adolescentes a partir de los 12 años. Se metaboliza a su metabolito activo (+)-O-desmetiltramadol mediante la enzima CYP2D6. La variabilidad genética en la actividad de CYP2D6 influye en la respuesta individual al tramadol.
Los metabolizadores lentostendrán concentraciones plasmáticas reducidas del metabolito, lo que afecta su efecto analgésico.
Los metabolizadores ultrarrápidosdebido a su elevada actividad metabólica experimentarán un incremento en las concentraciones plasmáticas de O-desmetiltramadol, lo que conlleva a una mayor incidencia de miosis (contracción excesiva de la pupila) y náuseas, así como de otros efectos adversos extremadamente peligrosos.
- VKORC1 y Warfarina
Las variantes en VKORC1 tienen un papel crucial en la respuesta individual a Warfarina, un anticoagulante oral ampliamente utilizado.
VKORC1 codifica la enzima vitamina K epóxido reductasa, que es esencial para la síntesis de factores de coagulación sanguínea dependientes de la vitamina K (como el factor II, VII, IX y X).
La Warfarina inhibe la actividad de VKORC1, reduciendo así la producción de factores de coagulación y previniendo la formación de coágulos.
El polimorfismo más estudiado en VKORC1 es la variante VKORC1 -1639G>A. se asocia con una mayor sensibilidad a la Warfarina por lo que la dosis debe ser disminuida, de lo contrario puede causar hemorragias severas.
- HLA y Abacavir
Los genes del complejo mayor de histocompatibilidad (HLA) están involucrados en la respuesta inmunitaria. Algunas variantes genéticas en estos genes se asocian a reacciones adversas graves a ciertos fármacos.
HLA-B*57:01 es una variante genética que está asociado con una mayor sensibilidad al abacavir, un fármaco utilizado en el tratamiento del virus de inmunodeficiencia humana (VIH). Se estima que entre el 8 % y el 61 % de los pacientes con VIH que portan esta variante y son tratados con abacavir desarrollarán hipersensibilidad al fármaco en un plazo de seis semanas.
Las personas con esta variante son más propensas a experimentar reacciones adversas como erupciones cutáneas.
- TPMT y Azatioprina
La enzima tiopurina metiltransferasa (TPMT) tiene la capacidad de descomponer una clase de medicamentos denominados tiopurinas, que incluyen azatioprina y mercaptopurina entre otras.
La azatioprina es un inmunosupresor se utiliza en trasplantes de órganos (riñón, corazón, hígado), enfermedades autoinmunes como artritis reumatoide y lupus, entre otras.
La mayoría de las personas no tienen dificultades para metabolizar las tiopurinas. Sin embargo, un pequeño porcentaje (aproximadamente 1 de cada 400) carece casi por completo de la capacidad para descomponer estos fármacos. Las personas sin actividad enzimática de la TPMT pueden experimentar efectos secundarios graves, como infecciones, anemia y sangrado, si reciben dosis normales de fármacos que contienen tiopurinas. En estos casos, los niveles de toxicidad del fármaco se acumulan en el organismo.
- UGT1A1 e Irinotecan
La UDP-glucuronosil-transferasa 1A1 (UGT1A1) es una enzima que tiene la capacidad de descomponer fármacos y también de eliminar una sustancia llamada bilirrubina del cuerpo.
El irinotecan se utiliza en el tratamiento sistémico del cáncer colorrectal metastático. Este fármaco es un inhibidor selectivo de la topoisomerasa I, una enzima crucial en la replicación celular. Es un profármaco y requiere conversión metabólica para volverse activa. La inactivación ocurre cuando se conjuga con la enzima UGT1A1, y el metabolito resultante se elimina por vía biliar y urinaria.
Una variante en la región responsable de iniciar la expresión del gen UGT1A1 afecta la actividad enzimática. La variante alélica UGT1A1*28, se asocia con menos actividad de UGT1A1. Esto resulta en una exposición prolongada al metabolito activo de Irinotecan, lo que conlleva a una mayor incidencia de efectos adversos graves con la dosis estándar de irinotecán (un riesgo aproximadamente de cuatro a cinco veces mayor de neutropenia grave y aproximadamente dos veces mayor de riesgo de diarrea grave).
En población caucásica, alrededor del 10% de los pacientes tienen esta variante. Estos pacientes tienen un mayor riesgo de toxicidad inducida por el tratamiento con Irinotecan, por lo que se debe reducir la dosis inicial del fármaco.
- VEGF y bevacizumab
El factor de crecimiento endotelial vascular (VEGF) es crucial para la formación de nuevos vasos sanguíneos (angiogénesis y vasculogénesis), procesos relacionados con el desarrollo de tumores. Para inhibirlos, se utiliza el bevacizumab, un anticuerpo monoclonal altamente específico que bloquea la unión del VEGF a sus receptores. El objetivo es evitar el crecimiento de tumores y mejorar la eficacia de otros tratamientos. El gen VEGF-A es fundamental, y variantes en él afectan la respuesta al bevacizumab. Por ejemplo, la variante rs833061 se asocia con una mejor respuesta, mientras que el genotipo −1154AA tiene baja eficiencia y mayor toxicidad.
- ABCB1 y capecitabina
El gen ABCB1 codifica para la glicoproteína P y pertenece a la familia de bombas de eflujo. Su función es expulsar fármacos de las células, evitando así la acumulación de sustancias tóxicas en el interior. Los pacientes con la variante ABCB1*1 han mostrado mayor toxicidad al tratamiento con capecitabina, un fármaco utilizado en el cáncer. Además, la variante C1236T en el gen ABCB1 se ha utilizado como biomarcador para predecir qué pacientes tolerarán mejor una quimioterapia basada en 5-fluorouracilo o capecitabina. La sobreexpresión de ABCB1 en tumores se ha relacionado con la resistencia a fármacos, lo que afecta la eficacia de los tratamientos.
- CYP3A4 y ciclofosfamida
La ciclofosfamida, utilizada en el tratamiento del cáncer de mama, se metaboliza principalmente a través de la enzima CYP3A4. Los polimorfismos en el gen CYP3A4 pueden afectar la formación de metabolitos tóxicos y, por lo tanto, la respuesta al tratamiento. Por ejemplo, los pacientes con el polimorfismo en CYP3A4 han mostrado una mayor toxicidad al recibir ciclofosfamida. Además, la variabilidad de efectos tóxicos según la etnia también es relevante, siendo los afroamericanos los que experimentan una mayor toxicidad.
Indudablemente, como se ha destacado a lo largo de este escrito, las implicaciones de las variantes genéticas en la respuesta a los fármacos son de gran relevancia, sin duda la farmacogenética desempeña un papel fundamental en la investigación.
Farmacogenética en México
En nuestro país el Instituto Nacional de Medicina Genómica (INMEGEN) desempeña un papel crucial en el avance de la farmacogenética. El INMEGEN se dedica a la investigación del genoma humano, identificando variantes genéticas específicas de la población mexicana. Además, el Instituto está a la vanguardia en el uso de tecnologías avanzadas de secuenciación y análisis genómico, lo cual es importante para identificar los polimorfismos genéticos que influyen en el metabolismo de los fármacos.
El Instituto ha liderado varios proyectos pioneros en la medicina genómica en México, como el Estudio del Genoma de los Mexicanos, que proporciona un mapa detallado de las variantes genéticas en la población mexicana.
Además, el INMEGEN cuenta con Laboratorio de Diagnostico Genómico el cual está integrado por técnicos de alta especialidad y tecnología de vanguardia. Se ofrecen pruebas moleculares para el pronóstico, diagnóstico y seguimiento de enfermedades, a personas e instituciones con el propósito de contribuir a mejorar el estado de salud y la práctica médica.
Finalmente, la visión del INMEGEN es “ser el referente nacional e internacional de investigación, desarrollo de políticas públicas e innovación en la salud preventiva. Sentando precedente de cómo la investigación en genómica puede tener un impacto directo en la toma de decisiones que cambien el panorama de las enfermedades que más afectan a México”.
Sin duda INMEGEN es fundamental para el progreso de la farmacogenética en nuestro país.
Conclusión
La farmacogenética representa un paso significativo hacia la medicina personalizada, donde los tratamientos se adaptan a las características individuales de cada paciente. A medida que esta ciencia avanza, podemos esperar una atención médica más eficaz, segura y centrada en el paciente. La promesa de la farmacogenética es clara: un futuro en el que cada persona reciba el tratamiento adecuado, basado en su perfil genético.
Referencias
- Rodríguez-Rojas, M., Moya, M., Martínez, A., & Gómez, M. (2018). Aplicación farmacogenómica de los genes CYP2C19, CYP2C9 y VKORC1 implicados en el metabolismo de los fármacos clopidogrel y warfarina. Revista de Cardiología, 24(4), 201–211. https://doi.org/10.1016/j.rccar.2018.05.005
- Dean L. Amitriptyline Therapy and CYP2D6 and CYP2C19 Genotype. 2017 Mar 23. Recuperado de: https://www.ncbi.nlm.nih.gov/books/NBK425165/
- Emery, L. P., & Brooks, G. A. (2022). Revisiting UGT1A1 Pharmacogenetic Testing Before Irinotecan-Why Not? JCO oncology practice, 18(4), 281–282. https://doi.org/10.1200/OP.21.00840
- EuroEspes Biotechnology. (n.d.). Farmacogenética. Recuperado de https://euroespes.com/farmacogenetica/
- Dean, L. (2018). Warfarin therapy and the VKORC1 and CYP genotype. https://www.ncbi.nlm.nih.gov/books/NBK84174/
- Cuervo del Pozo, L. (2022). Farmacogenética del cáncer. NPunto, V(56): 144-174. https://www.npunto.es/revista/56/farmacogenetica-del-cancer
- Instituto Nacional de Medicina Genómica. https://www.inmegen.gob.mx/
Lectura recomendada
- González Covarrubias, V. Farmacogenética y Farmacogenómica. ¿Qué es? ¿Nos sirve a todos? ¡Definitivamente! https://www.sabermas.umich.mx/archivo/la-ciencia-en-pocas-palabras/102-numero-13101/203-farmacogenetica-y-farmacogenomica.html
Fuente: acmor.org